Hematology
Authors: Agnes Davenport FNP-C and Nichole LePage, MD, Indiana University School of Medicine
Summary of clinical history
The patient is a 28-year-old female with a history of systemic lupus erythematosus (SLE) complicated by nephritis and severe cutaneous manifestations managed by multiple immunosuppressant medications. She was transferred to the emergency department after outpatient rheumatology labs revealed hemoglobin of 5.9 GM/dL [12-15] and platelet count of 10 k/mm3 [150-450].
Patient had a known history of iron deficiency anemia, menorrhagia and pancytopenia. She was admitted by the hematology service to undergo evaluation for severe anemia and thrombocytopenia. Her clinical examination was unremarkable.
| Measurement | Patient | Reference Range / Expected Result |
|---|---|---|
| Hemoglobin | 5.9 GM/dL | 12.0-15.0 |
| Platelet | 10 k/mm3 | 150-450 |
| MCV | 95fL | 81-99 |
| Reticulocyte | 5.2% | 0.5-2.5 |
| Haptoglobin | <6 mg/dL | 30-200 |
| Serum ferritin | 157.9 ng/mL | 10-106 |
| Serum Iron | 64mCg/dL | 50-212 |
| Total Iron-Binding Capacity | 260mCg/dL | 250-400 |
| PT | 12.2 seconds | 9.3-13.5 |
| INR | 1.03 | 0.73-1.17 |
| LDH | 773 U/L | 140-271 |
| Creatinine | 1.02 mg/dL | 0.6-1.2 |
| Folate | >23 ng/mL | >5.9 |
| Vitamin B12 | 151pg/mL | 200-1000 |
| DAT | Negative | Negative |
Microscopic findings
A peripheral blood smear was consistent with laboratory findings of anemia and thrombocytopenia. Abnormalities of red blood cell morphology were appreciated including dacrocytes, macrocytes, microcytes, and ovalocytes. There were many schistocytes appreciated on the sample collected on the day of admission. Schistocytes in combination with an elevated lactate dehydrogenase are consistent with microangiopathic hemolytic anemia.
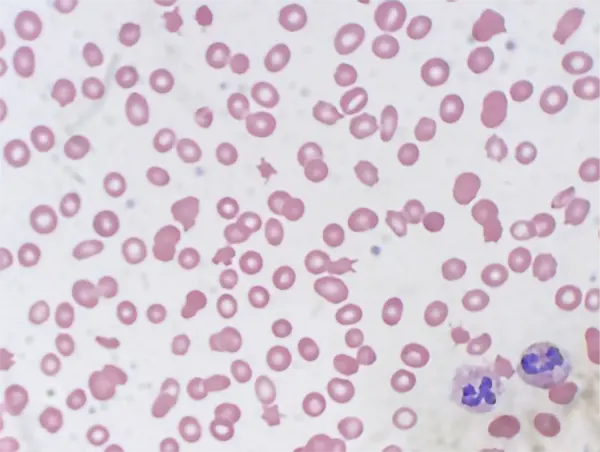
Clinical Course
Patient was started on corticosteroids due to concerns of immune-related pancytopenia etiologies and folinic acid for possible methotrexate toxicity. She was noted to have vaginal bleeding consistent with her known history of menorrhagia and this could have contributed to her anemia in addition to hemolysis. Tests for anticardiolipin and anti-beta-2-glycoprotein antibodies were below clinically significant thresholds, her serum methotrexate level resulted at <0.03 mCmol/L, and her ADAMTS13 activity resulted at <5%.
Neurological and physical exam remained within normal limits. Her platelet count remained below 20 k/mm3 despite two platelet transfusions. Her anemia responded appropriately to red blood cell transfusions despite persistent heavy vaginal bleeding.